ROLE OF PHYSISORPTION STATE IN MOLECULAR SCATTERING: A SEMI-LOCAL DENSITY-FUNCTIONAL THEORY STUDY ON O2/AG(111)
Molecular scattering experiments at surfaces, which are very sensitive to the topology of the interaction potential, have traditionally been considered as a reliable technique to indirectly confirm the existence of physisorption and chemisorption states.
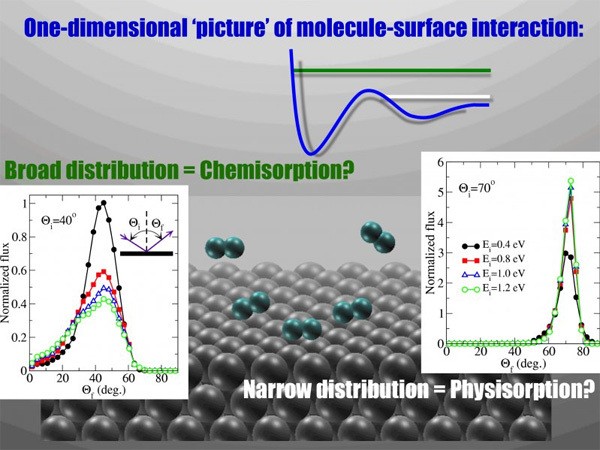
The work by Goikoetxea et al. that has recently been published in Physical Review Letters cast severe doubts on this still prevalent notion that dates back to the pioneering study by Sir Lennard-Jones [Trans. Faraday Soc. 28, 333 (1932)] and is based on the simplified one dimensional interaction potential picture he proposed at that time.
Using state-of-the-art gas/surface simulations, the authors have studied the scattering properties of O2 on Ag(111). This system has long been considered as a prototypical example of the scattering properties originated by the co-existence of repulsive walls arising from a physisorption and chemisorption part of the interaction potential. Nonetheless, simulations performed by Goikoetxea et al. on a six-dimensional potential energy surface that is calculated within semi-local density functional theory and, therefore, does not feature any physisorption well, can fully reproduce all the peculiarities of the scattering data. This work demonstrates that the existence of different repulsive regions in the potential energy surface must not necessarily imply the existence of a separate physisorption state, as has been traditionally assumed when interpreting the data in molecular scattering experiments.