Ultrafast Transient Dynamics of Adsorbates on Surfaces Deciphered
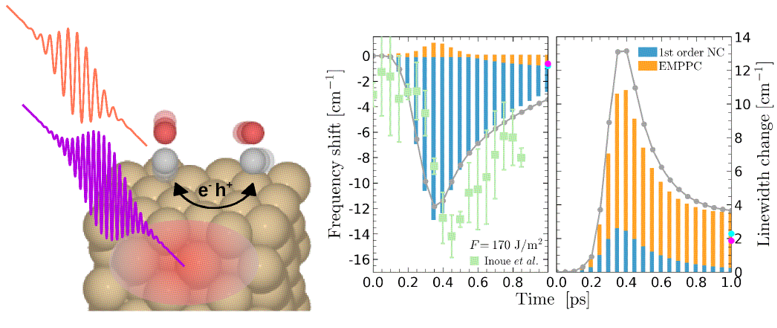
FIGURE: (Left panel) Schematic image of the coupling mechanisms underlying the internal stretch frequency shifts of CO on Cu(100) induced by femtosecond laser pulses. Contribution-resolved analysis of (middle panel) frequency shifts and (right panel) linewidth changes at an absorbed laser fluence F= 170 J/m2. Blue bars represent the contribution coming from the first-order nonadiabatic coupling (NC) term, orange bars show the electron-mediated phonon-phonon coupling (EMPPC) contribution, and gray circles are the sums of the two. Light blue and magenta points show the corresponding results for t= 50 and 100 ps, respectively. Green squares are the frequency shifts measured by K.-i. Inoue, et al. (Phys. Rev. Lett. 117, 186101 (2016)).
Probably one of the most exciting yet challenging goals in surface chemistry is to achieve control over surface reactions in both time and space domains. To reach this aim, many pump-probe spectroscopy techniques have been utilized for monitoring elementary motions and reactions of adsorbates on metal surfaces, including vibrations, diffusion, and desorption.
In this letter, Novko et al. take an important step towards this goal by developing a quantitative theoretical framework that is able to disentangle the contributions of electron- and vibration-driven interaction channels in ultrafast vibrational motion of molecular adsorbates on metal surfaces under nonthermal conditions.
As an example, the authors investigate the subpicosecond dynamics of the internal stretch mode of CO adsorbed on Cu(100) induced by femtosecond laser pulses. Their calculations perfectly reproduce the transient frequency and linewidth changes that were measured in time-resolved sum-frequency spectroscopy experiments to track how the laser-induced hot-carriers initiate a specific adsorbate reaction, CO desorption in this case. However, in contrast to the current understanding, the authors demonstrate that the frequency and linewidth changes bring information on two distinct mechanisms. Frequency shifts are caused by the nonadiabatic coupling between the laser-excited surface electrons and the internal stretch mode, while electron-mediated vibrational mode coupling controls the linewidth changes. In fact, this latter coupling turns out to be even richer than usually expected, since different modes dominate the ultrafast CO dynamics within the femtosecond and picosecond scales, as their calculations reveal. All in all, the in-depth understanding that this new theoretical framework can provide will be fundamental not only to the development of vibrational spectroscopy at surfaces, but additionally to advance in our ultimate goal of controlling surface reactions at the atomic level.