Understanding the Photoinduced Desorption and Oxidation of CO on Ru(0001) Using a Neural Network Potential Energy Surface
Upon femtosecond pulse laser irradiation on the Ru(0001) surface with coadsorbed CO and O, both CO desorption and oxidation take place. Using a Neural Network Potential Energy surface to perform molecular dynamics simulations, experimental results are reproduced and the mechanisms governing the underlying ultrafast photochemical processes are revealed.
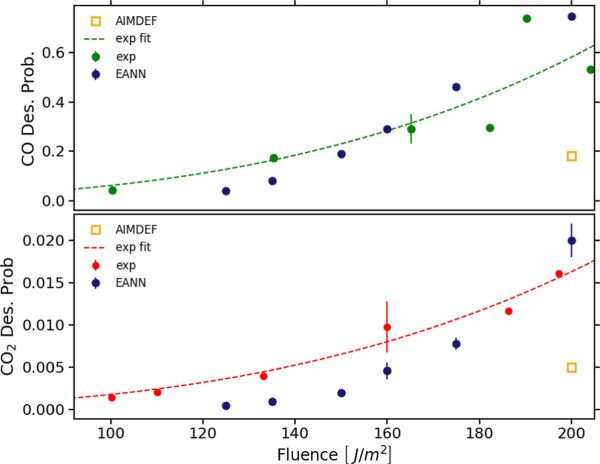
Figure 1. Fluence dependence of CO (top panel) and CO2 (bottom panel) desorption probabilities upon irradiating the CO/2O/Ru(0001) surface with an 800 nm laser pulse of fwhm 110 fs. Green (top panel) and red (bottom panel) circles are the experimental results for CO and CO2 desorption, respectively. Dashed lines in both panels are obtained by fitting the experimental data and should be used to guide the eye. Blue circles and error bars (the latter masked in many cases by the symbol size) are the results of the NNPES-based molecular d dynamics simulations with 30-50 ps simulation time. Open orange squares are the abinitio molecular dynamics results for F = 200 J/m2 with 4 ps simulation rtime.
CO oxidation on Ru(0001) is a reaction that, being thermally forbidden in ultra-high vacuum, can be activated by femtosecond laser pulses. Still, it is observed that CO desorption is much more likely than CO2 desorption with a branching ratio of around 35. Examining these experiments requires conducting molecular dynamics simulations in an excited environment. This can be performed at the level of density functional theory (DFT), by means of the so-called ab initio molecular dynamics with electronic friction and thermostats. In this approach, a set of coupled Langevin equations of motions are solved for the adsorbates and lattice atoms coupled to the electrons excited by the laser pulse, calculating the adiabatic forces on the fly using the Hellman-Feynman theorem. A key limitation of this approach is its substantial computational demand that, in practice, means that only a few hundred of trajectories restricted to an integration time of few picoseconds can be computed for specific experimental conditions.
In the present work, using the embedded atom neural network method, a multidimensional neural network potential energy surface (NNPES) is constructed, trained on configurations extracted from previous abinitio molecular dynamics simulations. This allows to perform simulations for different laser fluences F in the experimental range F=125-200 J/m2. The number of trajectories (2500-7500) and integration time (30-50 ps) vary for each fluence to ensure statistically accurate and time-converged results, allowing us a quantitative comparison with the experimental data. The calculated probabilities align remarkably well for both CO and CO2 desorption with the experimental data. First, the large branching ratio for CO desorption over CO2 desorption is reproduced. Second, both probabilities clearly exhibit the usual nonlinear dependence on F that characterizes photoinduced processes involving multiple electronic excitations. More importantly, the simulations are able to reproduce quantitatively the experimental data.
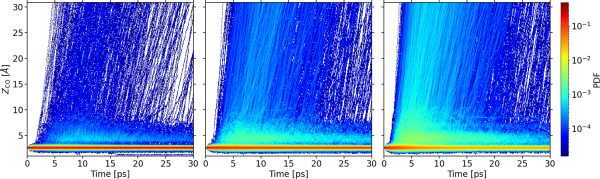
Figure 2. Normalized probability density of the CO center of mass height ZCO as a function of time for three different absorbed laser fluences: 125 J/m2 (left), 160 J/m2 (middle), and 200 J/m2 (right). ZCO is measured from the mean position of the Ru topmost layer. Bin area for all figures is 0.01 Å ps.
Finally, by analyzing the movement of the molecules along the dynamics, it is found the existence of dynamic trapping of CO that, in some cases, acts as a precursor for CO desorption. The simulations show that the energy landscape seen by the desorbing CO under equilibrium conditions and in the highly excited environment created by the laser pulse are drastically different, suggesting that the observed dynamic trapping is a consequence of the strong distortions and concomitant complex interactions created in the system.
All in all, the constructed NNPES is a powerful tool to describe at the DFT level the dynamics of the CO/O/Ru(0001) system under strong excitation conditions and makes it possible to perform statistically full converged realistic molecular dynamics simulations of complex laser induced processes that involved highly excited electronic and phononic sytems.