Why ultrafast photo-induced CO desorption dominates over oxidation on Ru(0001)
CO oxidation on Ru(0001) is a long-standing example of a reaction that, being thermally forbidden in ultra-high vacuum, can be activated by femtosecond laser pulses. In spite of its relevance, the precise dynamics of the photo-induced oxidation process as well as the reasons behind the dominant role of the competing CO photo-desorption have so far remained unclear.
Irradiation of a metal surface with femtosecond laser pulses generates a transient nonequilibrium distribution of hot electrons that subsequently transfer their energy to the surface phonons and also to the adsorbate degrees of freedom. This strong perturbation can lead to the emergence of completely new phenomena such as the opening of new reaction channels that cannot be accessed by thermal activation.
The oxidation of CO on Ru(0001) is precisely the emblematic example of a reaction that, being thermally forbidden in ultra-high vacuum, can be activated by femtosecond laser pulse irradiation, as was first found experimentally in Bonn et al. Science 285, 1042 (1999). Nonetheless, in spite of its relevance, the precise dynamics of the photo-induced CO oxidation process as well as the reasons behind the surprising dominant role of the competing CO photo-desorption process have remained unclear.
In this work, Tetenoire and co-workers investigate these two reactions with ab initio molecular dynamics with electronic friction that account for the non-equilibrated and highly excited electrons and phonons created by the laser. Their simulations successfully reproduce the main features observed in the experiments such as the photo-desorption of CO and CO2, the large CO desorption to CO oxidation branching ratio, and the changes in the O K-edge X-ray absorption spectra attributed to the initial stage of the CO oxidation process. Importantly, the authors are finally able to monitor step by step how the ultrafast CO desorption and CO oxidation occur in the highly-excited system and explain why CO desorption dominates over the energetically favored oxidation. It is the O adsorbed at the fcc sites that primarily recombines with the adsorbed CO, following basically the intermediate extreme states of the minimum energy oxidation path. The reason behind the unexpected inertness to the otherwise energetically favored oxidation is two-fold: (i) the difficult access to the transition state region, that requires the O atom crossing the bridge site and finding the CO conveniently close and tilted to form the chemisorbed bent CO2 and (ii) the fact that this access does not guarantee a successful recombination.
Figure 1: Art image of the recombinative desorption of CO2 induced by femtosecond laser pulses
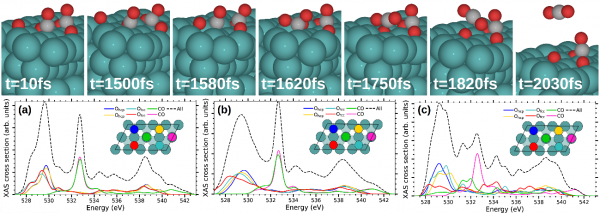
Figure 2: Top- Snapshots of a representative CO oxidation dynamics obtained in the (Te,Tl)-AIMDEF simulations (blue, red, and gray spheres correspond to Ru, O, and C atoms, respectively). The AIMDEF simulation time is indicated in each panel. Bottom- Time averaged O-K XAS cross section calculated at characteristic time intervals during the oxidative desorption process in the selected trajectory: (a) initial strong excitation of the adsorbates (0-1250 fs), (b) access to the transition state ruling the reaction, in which the recombining Ofcc reaches the bridge site that separates it from the nearest CO (1250-1580 fs), and (c) formation of the chemisorbed bent CO2 (1600-1630 fs). Each color curve shows the contribution of the corresponding colored O atom depicted in the surface unit cell plotted as an inset. Black dashed curves show the total time-averaged XAS.