Unified Understanding of the Structure, Thermodynamics, and Diffusion of Single-Chain Nanoparticle Fluids
Single-chain nanoparticles exhibit protein-like tunable architectures and versatile applications. Combining simulations and theory, we link their internal structure to anomalous dynamics and thermodynamics, revealing correlation holes, exponential scaling of properties, and weak caging. Universal trends emerge with implications for soft nanomaterials and glass formation.
Single-chain nanoparticles (SCNPs) are soft nano-objects synthesized via intramolecular cross-linking of polymer chains. Their tunable architectures—ranging from globular to sparse, akin to proteins—endow them with remarkable deformability and versatile functionalities, making them promising for applications in protein condensates, nanocomposites, nanomedicine, bioimaging, catalysis, and drug delivery.
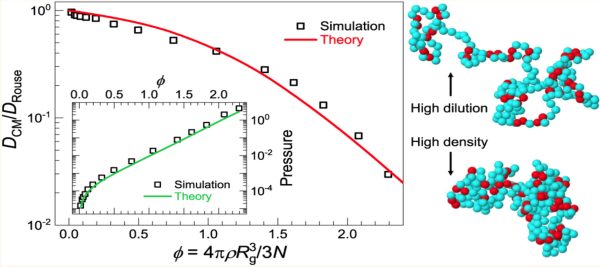
Figure. Left: Rouse-normalized SCNP diffusivity (main plot) and pressure (inset) vs. macromolecular packing fraction. Simulation results (symbols) are in excellent agreement with the theory (lines). Right: Typical conformations of a sparse (disordered protein-like) SCNP at low and high concentration. Red beads are cross-linked monomers.
To unravel their dynamic and thermodynamic behavior, we combine molecular dynamics simulations with a generalized Langevin equation theory, capturing mode-coupling effects and weak caging in SCNP fluids. Our framework bridges microscopic conformations to macroscopic properties, elucidating how internal SCNP structure governs intermolecular packing, thermodynamics, and center-of-mass diffusion across concentrations—from dilute solutions to dense melts.
Key findings reveal a distinctive correlation hole in intermolecular pair correlations, arising from SCNP connectivity and repulsive interactions at macromolecular scales. Concentration-dependent deviations emerge at small separations, while theoretical predictions—confirmed by simulations—uncover anomalous exponential-like scaling of pressure, osmotic compressibility, and diffusivity with packing fraction. Despite system-specific variations tied to internal architecture, universal trends emerge, rationalized through an effective globule model at macromolecular scales. The pronounced diffusivity decline with concentration stems from a nonactivated, excluded-volume-driven weak caging process, mediated by spatiotemporal intermolecular forces.
Our theory aligns closely with simulations and experimental viscosity data, offering testable predictions. The approach is extensible to other soft nanoparticles with intricate internal organization, paving the way for studies of kinetic arrest in macromolecular glasses.