Molecular doping in the organic semiconductor diindenoperylene: insights from many-body perturbation theory
In this study we use the GW/BSE (GW+ Bethe-Salpeter equation) approach to study the effect of molecular doping on organic semiconductors. Our results highlight the importance of accurately taking into account the solid-state screening and dopant-host interactions in order to satisfactorily describe the optoelectronic properties of those systems.
In this study we take advantage of the quantitative accuracy of the GW/BSE approach to describe the electronic structure and optical properties of an interesting organic semiconductor (OSC): diindenoperylene (DIP) molecular crystal. We focus on the doping of DIP crystals with acceptor molecules in order to control their carrier density and mobility. This is crucial to tune the conductivity and optical properties of this material for specific applications. Our results clearly indicate that, contrary to the customary belief, neither the information obtained from the isolated molecules nor from molecular crystals in the pure phase is sufficient to determine relevant dopant-host combinations. The interaction and hybridization with the host environment, including many-body effects, must be carefully considered in order to successfully identify appropriate molecular dopants for a given OSC. Fortunately, our paper demonstrates that the accuracy of current GW/BSE methods can be sufficient to allow identifying such dopants.
We first show that our theoretical estimates of the transport and optical gaps, as well as the optical absorption spectra, of the isolated and the crystalline DIP are in excellent agreement with available experimental data. We then perform a systematic analysis of the dopability of the DIP with two strong acceptors, namely F4TCNQ and F6TCNNQ molecules. Our analysis demonstrates that the studied doped crystals feature hybridized states at the valence-band edge associated with a host-dopant charge-transfer complex that must be taken into account in order to understand the electronic and optical properties of the system. Additionally, electrostatic and solid-state screening effects are instrumental in accurately describing the level alignment between dopant and host molecules.
In summary, we propose two new doped DIP crystals with salient features in their optical structures that could be exploited for optoelectronic applications. Furthermore, our results disprove the belief that the ionization energy and the electron affinity computed in the gas phase or even measured on films of pure host and dopant molecules can be applied to discuss the dopability of an OSC. In contrast, we demonstrate that environmental screening and interaction effects must be carefully taken into account to obtain a reliable picture of the doping of molecular crystals.
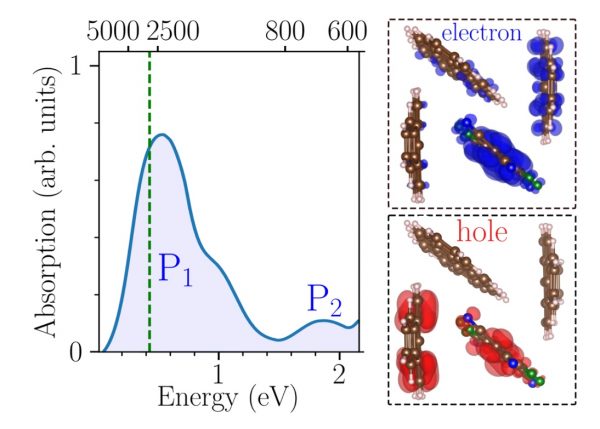
Figure: Left: Low energy optical absorption spectrum of the DIP crystal substitutionally doped with F6TCNNQ. Right: Scheme showing the unit cell containing three DIP molecules and one F6TCNNQ molecule, together with the spatial distribution of the hole/initial-state (red) and electron/final-state (blue) densities for the P1 peak. Note that the hole density is mainly centered on the HOMO of one of the host DIP molecules, while the electron density shows a large contribution from the LUMO of the dopant molecule. In spite of the hybridization between neighboring molecules, this highlights the dominant host-dopant charge-transfer character of the P1 transition.