Photoinduced desorption dynamics of CO from Pd(111): a neural network approach
Using the Embedded Atom Neural Network (EANN) Serrano-Jiménez et al. develop an accurate and extremely complex Potential Energy Surface (PES) that allows a detailed and reliable description of the photo-induced desorption of CO from Pd(111). The obtained EANN-PES is able to describe an extensive range of surface temperatures (90-1000 K), a large amount of degrees of freedom, and the varying CO coverage caused by the abundant desorption events.
Irradiation of an adsorbate-covered metal surface with femtosecond laser pulses, from the near-infrared to the ultraviolet regime, generates a transient nonequilibrium distribution of hot electrons and a subsequent excitation of the lattice atoms via electron-phonon coupling. As a result, adsorbates encounter a combined electronic and phononic excited system from which they can gain energy, facilitating the initial vibrational excitation, the subsequent diffusion on the surface, and the final recombinative desorption. Usually, the cross-sections for some of these photo-induced reactions are significantly increased respect to what it is observed under ordinary thermal excitation conditions.
From the theoretical side, the modelling of these experiments requires to perform multidimensional Molecular Dynamics (MD) simulations involving multiple adsorbates and surface atoms in the presence of nonequilibrium distributions of excited electrons and phonons. Only very recently this complete description of the problem has been achieved by means of the ab initio molecular dynamics with electronic friction and thermostats [(Te,Tl)-AIMDEF] model [M. Alducin et al. PRL 123, 246802 (2019)].
However, the extremely computationally demanding (Te,Tl)-AIMDEF methodology severely limits the achieved statistics, the simulation time, and the size of the system to model. These limitations could nowadays be overcome if it were possible to construct a multidimensional potential energy surface of ab initio quality, from which the atomic forces could be rapidly, but accurately, calculated. In principle, multidimensional potential energy surfaces can be generated using Neural Networks (NN-PES). However, the requirements imposed to a NN-PES capable of describing femtosecond laser induced reactions are unprecedentedly extreme and demanding due to the necessity of modeling the movement of multiple adsorbates and great number surface atoms under an ample range of varying lattice and electronic temperatures.
In this work, the authors demonstrate that the new embedded atom neural network method [Y. Zhang et al. J. Phys. Chem. Lett. 10, 4962 (2019)] allows to develop such accurate and extremely complex NN-PES for the specific case of the description of the photo-induced desorption of CO from the Pd(111) surface with a coverage of 0.75 monolayers. MD simulations performed in this NN-PES reproduce with great accuracy the ab initio molecular dynamics simulations. Moreover, the presented methodology is completely general and can be applied to any other adsorbate-surface system and different coverages. For all these reasons, it opens the path to, from a theoretical point of view, fully characterize and understand the femtosecond laser pulse induced chemistry at surfaces.
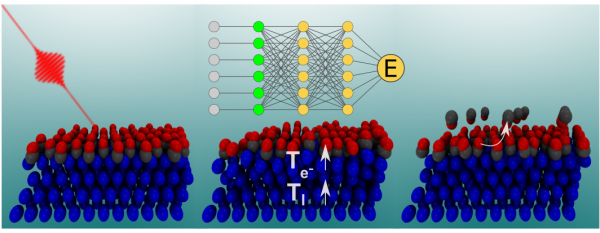
Figure: Scheme of the photo-induced desorption of CO from Pd(111) with a coverage of 0.75ML. Molecular dynamics simulations are performed using a Multi-Dimensional Neural Network Potential Energy Surface that reliably describes highly varying surface temperatures (90-1000 K) and CO coverages (0.33-0.75 ML).