Quantum corrections in the dynamics of hydrogen molecules at surfaces
Static properties of surfaces are in general well known. Computational resources have become powerful enough to describe them using first principles and there is extensive output extracted from experimental measurements on their electronic, magnetic, and optical properties. Now, the challenge is the atomic- and molecular-level understanding of surface dynamics.
Elementary processes arising in gas/solid interfaces are ever-present in our daily life as well as in many industrial applications. The oxidation and corrosion of surfaces, the catalytic converters used in cars, the surface doping of semiconductors, or the industrial production of most synthetic compounds are all of them relevant examples of the importance of surface physical and chemical processes.
Synergetic advances in theory and molecular beam experiments associated with a tremendous increase of computing power have allowed a detailed understanding of the dynamics of chemical elementary processes. For gas phase reactions involving a small number of degrees-of-freedom, quantum dynamics calculations might be now regarded as exact because they generally provide results in excellent agreement with experiments. However, the complexity of surface reaction dynamics prevents an efficient and even effective use of quantum dynamics. A crucial issue is thus to develop accurate semiclassical tools able to make realistic predictions when major quantum effects come into play in the dynamics.
In this work, the authors improve over a classical trajectory method by including quantum corrections. The interaction between the molecules and the surface are described by density functional theory. They assign statistical weights to classical paths on the basis of two semiclassical corrections: Gaussian binning and the adiabaticity correction. This approach was previously applied by them to the heterogeneous gas–surface reaction between H2 in its internal ground state and the Pd(111) surface [Rodríguez-Fernández et al., J. Phys. Chem. Lett., 2019, 10, 7629]. Its predictions of the sticking and state-resolved reflection probabilities were found to be in surprisingly good agreement with those of exact quantum time-dependent calculations where standard quasi-classical trajectory calculations failed. This subsequent work shows that the quality of the calculations is kept when the incident H2 molecules are rotationally excited, a feature extremely difficult to describe accurately.
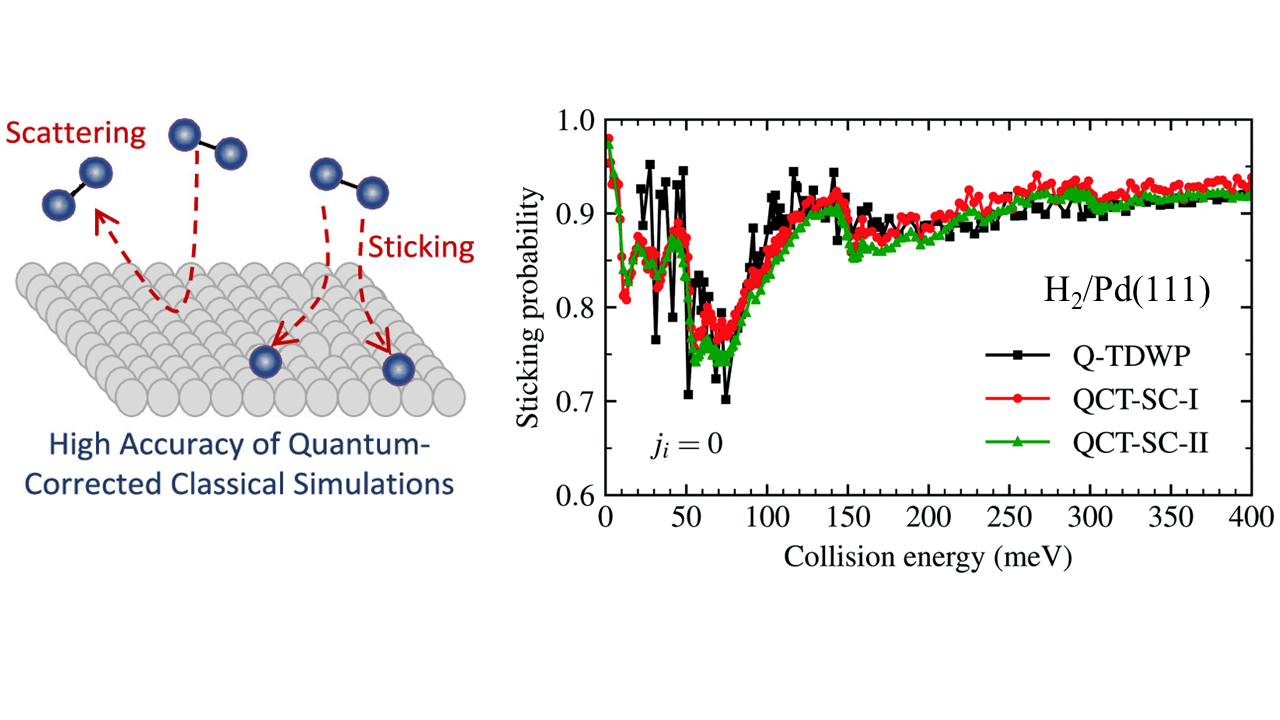
Figure: Sticking probability for H2 on Pd(111) as a function of the collision energy. Black symbols are purely quantum calculations. Red and green symbols represent quantum-corrected classical calculations.