Surface electron density models for accurate ab initio molecular dynamics with electronic friction
The interaction of thermal atoms and molecules with surfaces and nanostructures are ever-present in our daily life as well as in many industrial applications. The oxidation and corrosion of surfaces, the catalytic converters used in cars, or the industrial production of most synthetic compounds are relevant examples of the importance of the elementary reaction processes arising in gas/surface interfaces. Within this context, the challenge in present computational surface science is to count with reliable theoretical models that can predict the outcome of a specific reaction.
In the last decade, the ever more powerful computational resources have permitted us to advance in our understanding of gas-surface reactivity from first principles. There are many theoretical studies confirming that the fundamental properties in most elementary gas-surface processes are satisfactorily described by the Born-Oppenheimer approximation, which neglects the coupling between electronic excitations and the nuclear motion. Still, the challenge in present gas-surface simulations is to provide a reliable description of the two main energy exchange channels that may affect the dynamics and reactivity of atoms and molecules on solid surfaces, namely, phonons and electron-hole (e-h) pair excitations. In the end, these are the mechanisms that dictate, on the one side, the thermalization rate and, hence, the mean traveled the length of the nascent adsorbates, and, on the other side, the mean traveled path of the desorbing species. It is the balance between all these key quantities that will determine the catalytic properties of the surface.
In order to tackle this issue, the gas-surface dynamics group at CFM has developed the so-called ab initio molecular dynamics with electronic friction (AIMDEF) scheme that provides a joint and reliable description of the effect that e-h pairs and phonons will have on elementary reactions at metals. Using this new AIMDEF method to study the relaxation dynamics of different atoms and molecules, the authors are able to extract general trends on the role and competition between both energy exchange channels: (i) e-h pairs are expected to dominate the dynamics of light gas species and (ii) even if phonons will increasingly dominate the heavier hot species, the contribution of e-h pairs is by no means negligible in these cases because it gains relevance at the last stage of the relaxation process.

Figure 1. Schematic representation of electron-hole pair excitations during the dissociative adsorption of H2 on Pd(100)
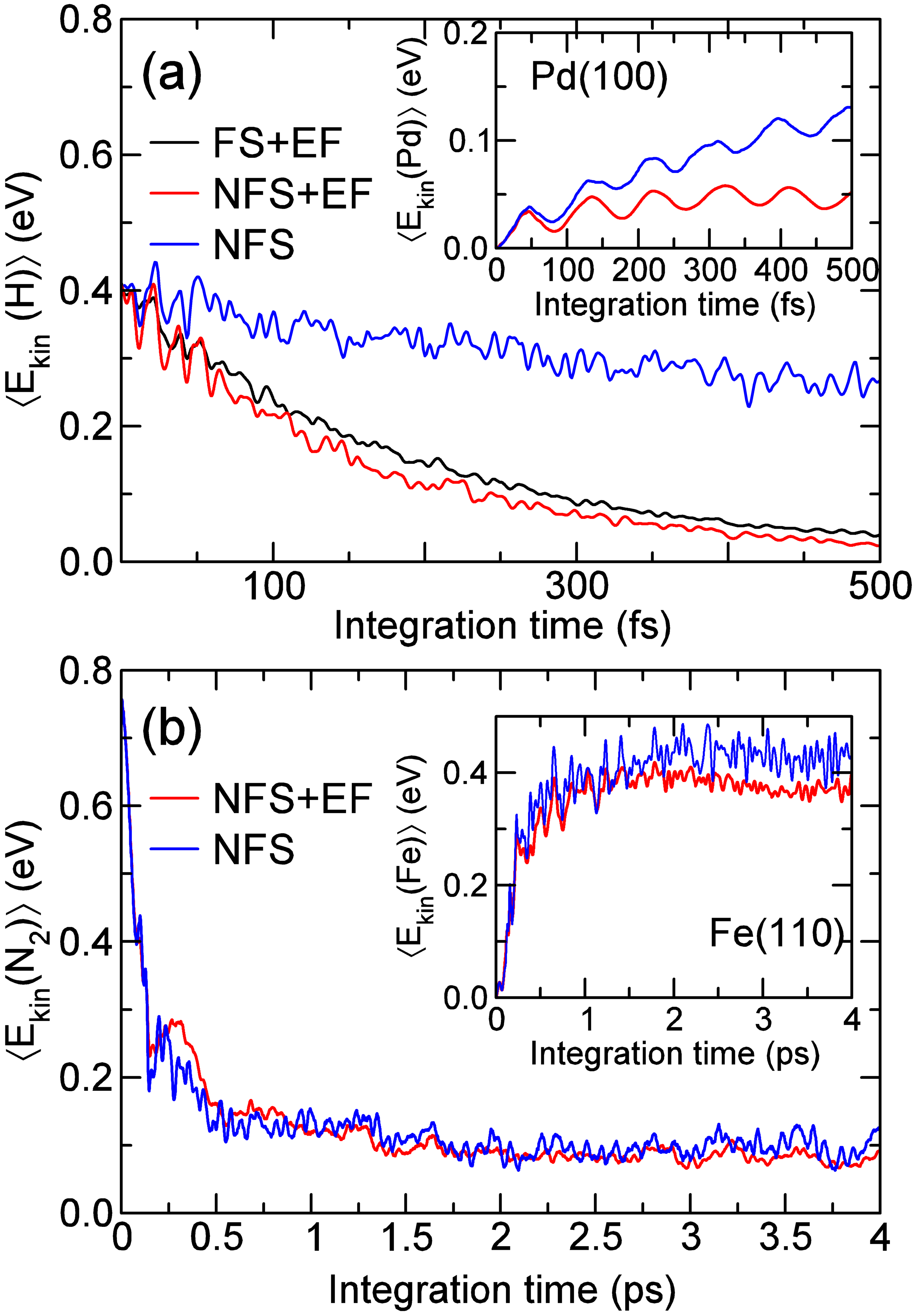
Figure 2. (a) Time evolution of the mean kinetic energy of the nascent H atoms formed upon dissociation of 0.5 eV H2 on Pd(100). (b) Same for 0.75~eV N2 adsorbing on Fe(110). Results obtained from different AIMDEF simulations: including both e-h pairs and phonons (red curves, labeled NFS+EF), only phonons (blue curves, labeled NFS), and only e-h pairs (black curve, labeled FS+EF). Each inset shows the mean kinetic energy of the surface atoms versus time.