dynamics and reactivity of gas-phase molecules at surfaces
1 / 4

dissociating dynamics
2 / 4
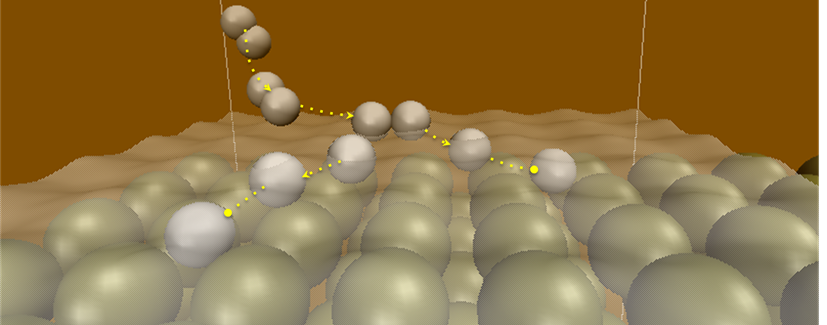
non-adiabatic dynamics
3 / 4
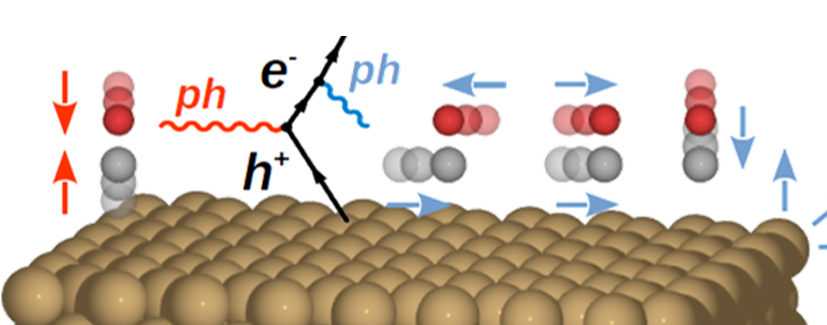
vibrational lifetime of adsorbates
4 / 4

photo-induced reactions
In general terms, understanding and mastering the physics and chemistry of
adsorption processes at nanostructures and surfaces is a basic requirement
for the full development of nanoscience and nano-technology.
Metal surfaces are effective chemical agents capable of adsorbing and/or
dissociating molecules impinging from the gas phase. Industrial processes
of enormous economical impact, such as corrosion and heterogeneous catalysis,
greatly benefit from recent developments in basic research on this matter.
Over the last years, the combination of experimental molecular-beam techniques
and refined theoretical calculations based on ab-initio methods have
led research on the field to a new stage, in which detailed investigations
of the kinetics and dynamics of molecular reactivity at surfaces are possible.
In the "Gas/Solid Interfaces" group, we are mainly interested in the elementary reactive processes that may happen whenever atoms or small molecules interact with surfaces. When a molecule approaches the surface, intramolecular chemical bonds
can break down and new ones be formed with the surface.
We use first-principles electronic structure calculations to
describe the details of the interaction between the incoming species and the surface
through a multidimensional potential energy surface (PES). Once the PES of
the system is known, we simulate the dynamics of several processes by solving
the classical equations of motion of the nuclei.
We pay particular attention to the non-adiabatic processes and the energy dissipation channels that come into play, because they can drastically change the output of the dynamics.
From a theoretical point of view, the description of
ground state properties is currently well founded and has proven to be extremely successful in
explaining elementary reactive and non-reactive adiabatic processes at surfaces. The
description of excited states and the evaluation of energy transfer mechanisms is however still
maturing and further developments are needed to reach the same level of detail in the
understanding and of accuracy in the quantitative representation.
group members
Maite Alducin, tenured scientist, CFM CSIC-UPV/EHU
Ricardo Díez Muiño, Ikerbasque research professor, DIPC
J. Iñaki Juaristi, catedrático de universidad, CFM CSIC-UPV/EHU
Oihana Galparsoro, profesor adjunto de universidad, Facultad de Química, UPV/EHU
Alfredo Serrano, PhD student at the MPC and UPV/EHU
Ivan Žugec, PhD student at the MPC and UPV/EHU
Natalia Koval, postdoctoral researcher at CFM CSIC-UPV/EHU
external group members
Alberto Sánchez Muzas, profesor ayudante Dr. at Univesidad Autóma de Madrid
Raúl Bombín, postdoctoral researcher at Université de Bordeaux
financial support
Spanish MCIN/AEI/10.13039/501100011033/ [Grant No. PID2022-140163NB-I00]
Gobierno Vasco-UPV/EHU Project No. IT1569-22
Gobierno Vasco IKUR program
former group members
Auguste Tetenoire, PhD student at DIPC and UPV/EHU
Alberto Rodríguez, PhD student at UPV/EHU and Université de Bordeaux
Alejandro Peña, PhD student at UPV/EHU and Université de Bordeaux
Ivor Lončarić, PhD student at CFM, CSIC-UPV/EHU
Mohammed Ahmed Nosir, PhD student at CFM, CSIC-UPV/EHU
Dino Novko, PhD student at DIPC
Itziar Goikoetxea, PhD student at CFM, CSIC-UPV/EHU
Gisela A. Bocan, postdoctoral fellow at DIPC
Ludovic Martin-Gondre, postdoctoral fellow at DIPC
Geethalakshmi Rangaswa, postdoctoral fellow at CFM, CSIC-UPV/EHU
Alejandro Rivero, postdoctoral researcher at the DIPC
selected recent publications
Dynamic Training Enhances Machine Learning
Potentials for Long-Lasting Molecular Dynamics
I. Žugec, J. I. Juaristi, and M. Alducin
(preprint)
Strong-field effects in the photo-induced dissociation of the hydrogen molecule on a silver nanoshell
N. Koval, J. I. Juaristi, and M. Alducin
Chem. Sci. 15, 18581 (2024)
Understanding the Photoinduced Desorption and Oxidation of CO on Ru(0001) Using a Neural Network Potential Energy Surface
I. Žugec, A. Tetenoire, A. S. Muzas, Y. Zhang, B. Jiang, M. Alducin, and J. I. Juaristi
J. Am. Chem Soc. Au 4, 1997-2004 (2024)
Multicoverage Study of Femtosecond Laser-Induced Desorption of CO from Pd(111)
A. S. Muzas, A. Serrano Jiménez, Y. Zhang, J. , B. Jiang, J. I. Juaristi, and M. Alducin
J. Phys. Chem. Lett. 15, 2587-2594 (2024)
How Adsorbed Oxygen Atoms Inhibit Hydrogen Dissociation on Tungsten Surfaces
A. Rodríguez, L. Bonnet, P. Larregaray, and R. Díez Muiño
J. Phys. Chem. Lett. 14 (5), 1246-1252 (2023)
Why Ultrafast Photoinduced CO Desorption Dominates over Oxidation on Ru(0001)
Auguste Tetenoire, Christopher Ehlert, J. I. Juaristi, Peter Saalfrank, and M. Alducin
J. Phys. Chem. Lett. 13 (36), 8516-8521 (2022)
Photoinduced Desorption Dynamics of CO from Pd(111): A Neural Network Approach
A. Serrano Jiménez, A. P. S. Muzas, Y. Zhang, J. , B. Jiang, I. Lončarić, J. I. Juaristi, and M. Alducin
J. Chem. Theory Comput. 17, 4648-4659 (2021)
Absence of spillover of hydrogen adsorbed on small palladium clusters anchored to graphene vacancies
A. Granja-DelRío;, M. Alducin, J. I. Juaristi d,b, M-J. López, J. A. Alonso
Applied Surface Science 559, 149835 (2021)
When classical trajectories get to quantum accuracy: The scattering of H2 on Pd(111)
A. Rodríguez-Fernández, L. Bonnet, P. Larregaray, and R. Díez Muiño
Phys. Chem. Lett. 10, 7629 (2019)
Electrons and Phonons Cooperate in the Laser-Induced Desorption of CO from Pd(111)
Maite Alducin, Nicholas Camillone, III, Sung-Young Hong, and J. Iñaki Juaristi
Phys. Rev. Lett. 123, 246802 (2019)
Read also CFM-Highlight
Ultrafast Transient Dynamics of Adsorbates on Surfaces Deciphered: The Case of CO on Cu(100)
D. Novko, J. C. Tremblay, M. Alducin, and J. I. Juaristi
Phys. Rev. Lett. 122, 016806 (2019)
Read also CFM-Highlight
Read also Highlight at the Institute of Physics (Zagreb)
Electron-mediated phonon-phonon coupling drives the vibrational relaxation of CO on Cu(100)
D. Novko, M. Alducin, and J. I. Juaristi
Phys. Rev. Lett. 120, 156804 (2018)
Read also CFM-Highlight
Read also article at Mapping Ignorance
Electronic Stopping of Slow Protons in Oxides: Scaling Properties
D. Roth, B. Bruckner, G. Undeutsch, V. Paneta, A. I. Mardare, C. L. McGahan, M. Dosmailov, J. I. Juaristi, M. Alducin, J. D. Pedarnig, R. F. Haglund, Jr., D. Primetzhofer, P. Bauer
Phys. Rev. Lett. 119, 163401 (2017)
Strong anisotropic interaction controls unusual sticking and scattering of CO at Ru(0001)
Lončarić I, Fuchsel G, Juaristi JI, and Saalfrank P.
Phys. Rev. Lett. 119, 146101 (2017)
Non-adiabatic effects in elementary reaction processes at metal surfaces
M. Alducin, Díez Muiño, and J.I. Juaristi
Progress in Surface Science 92, 317 (2017)
Read also article at Mapping Ignorance
Electronic Stopping of Slow Protons in Transition and Rare Earth Metals: Breakdown of the Free Electron Gas Concept
D. Roth, B. Bruckner, M. V. Moro, S. Gruber, D. Goebl, J. I. Juaristi, M. Alducin, R. Steinberger, J. Duchoslav, D. Primetzhofer, P. Bauer
Phys. Rev. Lett. 118, 103401 (2017)
Read also DIPC-Highlight
Read also article at Mapping Ignorance
Surface electron density models for accurate ab initio molecular dynamics with electronic friction
D. Novko, M. Blanco-Rey, M. Alducin, and J.I. Juaristi
Phys. Rev. B 92, 245435 (2016)
Read also CFM-Highlight
Femtosecond-laser-driven molecular dynamics on surfaces: Photodesorption of molecular oxygen from Ag(110)
I. Lončarić M. Alducin, P. Saalfrank, and J. I. Juaristi
Phys. Rev. B 93, 014301 (2016)
Ab initio molecular dynamics with simultaneous electron and phonon excitations: Application to the relaxation of hot atoms and molecules on metal surfaces
D. Novko, M. Blanco-Rey, J.I. Juaristi, and M. Alducin
Phys. Rev. B (Rapid Comm.) 92, 201411 (2015)
Surface strain improves molecular adsorption but hampers dissociation for N2 on the
Fe/W(110) surface
I. Goikoetxea, J. I. Juaristi, R. Díez Muiño, and M. Alducin
Phys. Rev. Lett., 112, 156101 (2014)
Read also CFM-Highlight
Electronic friction dominates hydrogen hot atom relaxation on Pd(100)
M. Blanco-Rey, J. I. Juaristi, R. Díez Muiño, H. F. Busnengo, G.-J. Kroes, and M. Alducin
Phys. Rev. Lett., 112, 103203 (2014)
Read also CFM-Highlight
Role of physisorption states in molecular scattering: A semilocal density-functional
theory study on O2/Ag(111)
I. Goikoetxea, J. Meyer, J. I. Juaristi, M. Alducin, and K. Reuter
Phys. Rev. Lett., 112, 156101 (2014)
Read also CFM-Highlight
other representative references
Dynamics of gas/surface interactions: Atomic-level understanding of scattering processes at surfaces
Edited by R. Díez Muiño and H. F. Busnengo
'Springer Series of Surface Sciences' vol. 50 (Springer, Berlin 2013), ISBN 978-3-642-32954-8.
Efficient N2 formation on Ag(111) by Eley-Rideal recombination of hyperthermal atoms
M. Blanco-Rey, E. Díaz, G. A. Bocan, R. Díez Muiño, M. Alducin, and J. I. Juaristi
J. Phys. Chem. Lett. 4, 3704 (2013)
DOI: 10.1021/jz401850h
Competition between electron and phonon excitations in the scattering of nitrogen atoms and molecules off Tungsten and Silver metal surfaces
L. Martin-Gondre, M. Alducin, G. A. Bocan, R. Díez Muiño, and J. I. Juaristi
Phys. Rev. Lett. 108, 096101 (2012)
Role of electron-hole pair excitations in the dissociative adsorption of diatomic molecules on metal surfaces
J. I. Juaristi, M. Alducin, R. Díez Muiño, H. F. Busnengo, and A. Salin
Phys. Rev. Lett. 100, 116102 (2008).
Why N2 molecules with thermal energy abundantly dissociate on W(100) and not on W(110)
M. Alducin, R. Díez Muiño, H. F. Busnengo, and A. Salin
Phys. Rev. Lett. 97, 056102 (2006).